Edit: if you are working in a group of 3, include a workload breakdown (i.e. what each person will do).
The goal of the final project is to gain experience with the entire scientific process in bioinformatics: motivation, hypothesis development, data collection/cleaning, algorithm development/application, evaluation of results, interpretation, and conclusion. Below I'll describe the main components your project should include and give a few example projects. You are welcome to choose something outside of this list, but check with me first if it is outside the type of material we've been doing in this class.
This document describes the project proposal and getting started on the project - there will be separate instructions for the oral presentation and exactly what to submit at the end of the semester. The project is worth 15% of your overall grade.
For the oral presentations, each student will have roughly 4 minutes to speak (+ time for questions).
I would encourage you to work in pairs, but if you would prefer to work individually or in a group of 3, email me as soon as possible to arrange. Project expectations will scale linearly with the number of people in your group. You are welcome to work across lab sections (but we will still have required lab the last two weeks of classes and it would be ideal if both people could come to the same lab).
I am not doing formal random partners for the project, but if you would like to be matched with someone, send me an email and let me know if there were partners throughout the semester that you worked particularly well with.
The goal of the proposal is to help you start working on your final project and assembling the different resources you want to use (literature, software, data, etc). Broadly, your project should include:
For the proposal, briefly outline each of these sections (details below) in a 1-page PDF document. Print and submit your proposal to me in 260 (dropbox outside my office is fine if I'm not there). Include a title and both partner names.
Why is this an interesting or relevant topic? You could talk about your personal motivation for this topic, or why it would be interesting to investigate in general. What scientific question are you trying to answer? For example, your question could be: is BWA or Bowtie a more accurate read aligner? The answer could depend on exactly what dataset you investigate, what parameters you use, how you define accuracy. Briefly describe a hypothesis about the results you expect. (i.e. I think Bowtie will be more accurate because the back-tracing is more thorough, etc.)
What dataset will you use for this project? It could be real data or simulated data (if your ideal data is not available, simulated data is great alternative and I'm happy to talk more about that). Please be as specific as possible - don't assume your ideal data is available until you've actually downloaded and viewed it. If you are using real data, include a link to the data in your proposal. Here are a few databases to get started:
NCBI: Variety of data formats for thousands of different species (plants, animals, viruses, etc). Click on the "Downloads" tab. List by organism.
dbSNP: SNP variation data for humans (within the NCBI umbrella). I would probably recommend 1000 genomes or SGDP over dbSNP, but this database could be helpful for more health-related questions.
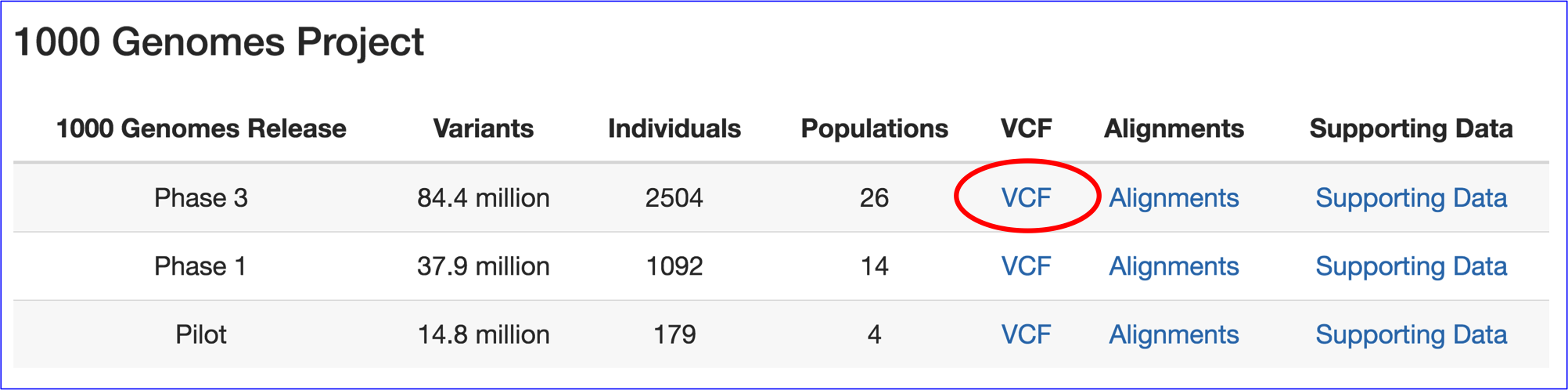
1000 genomes: More than 1000 humans genomes from around the world. There are fewer populations and more individuals per population (relative to the SGDP dataset). I would recommend using the first group of VCF files.

Neandertal genomes: Several ancient genomes, compared to human sequences.
Simons Genome Diversity Project (SGDP): This is a really nice dataset of human variation (more populations and fewer individuals per population relative to 1000 genomes). Notice that they use BWA to align the short reads! I would recommend working with the VCF files. The download is large (57G). If several groups want to work on this dataset, let me know and I'll put it somewhere convenient. (You typically won't need to use all the individuals or all the chromosomes.)
Tomato genomes: I highlight this dataset in particular since it is in a good format and includes both wild tomatoes (smaller, different colors) and the more common supermarket variety (selected to be bigger and redder). There could be some interesting population genetic analysis on this dataset. Domesticated crops (corn, rice, soybeans, etc) and animals (cattle, poultry, etc) also often have good public datasets.
Short Read Archive (SRA): this is a database of short reads (similar to what we used for genome assembly and read alignment). If your projects related to assembly or alignment, I would definitely recommend the short read archive, which has data from many different species. Note that most of these files are very large, so try to start small with fractions of the data or species with smaller genomes.
What software/methods will you use for the project? You can write your own method(s) or use existing software, but there should be some programming component. You could compare two existing methods, or compare your own method to an existing method. Even if you're planning to use existing software, there will likely be a significant programming component since you'll have to get the data in the right format, learn how to run the program, and evaluate the results.
If you are using any existing software, include a link.
What type of results do you expect from your project? How will you evaluate the results? What might you be able to say about biology at the end of the project?
Include a list of at least two references (Google scholar is a great place to start). If you would like paper recommendations based on your topic, let me know. For the reference format, include:
Example:
Langmead, Trapnell, Pop, et al. "Ultrafast and memory-efficient alignment of short DNA sequences to the human genome." Genome Biololgy (2009).
If your references include online references (software, datasets, etc) or books, feel free to modify this format (i.e. title of site and link is usually enough).
Here are a few high-level ideas to get you started. You are welcome to do something completely different, but check with me first if it is very far outside our class material. Some of these relate to topics we haven't covered yet, but I wanted to include them anyway. Many of these are method comparisons, which I like because they provide a concrete goal and allow you to work with state-of-the-art software. You are welcome to modify any of these projects to include a comparison with your own software. You can also investigate only one program/algorithm (either your own or existing) and experiment with modifications or different input parameters.
Genome Assembly. In class we looked at the Velvet assembler, but since its development many other assemblers have been released. A comparison of 2 or more of these methods would be very interesting. The Assemblathon 2 paper might be a good place to get started.
Multiple Sequence Alignment. In class we talked a lot about pairwise sequence alignment, but often we want to consider multiple sequences. There are several popular multiple sequence alignment algorithms one list here, and a comparison of 2 or more of these methods would be very interesting. Alternatively, you could investigate one multiple sequence alignment algorithm and then use the output as input for a phylogenetic tree algorithm.
Read Alignment. A comparison of BWA and Bowtie would be very interesting (in terms of either accuracy or speed). Since some comparisons exist already, I would recommend trying a new dataset (see the SRA dataset above).
Phylogenetic Trees. Many modern phylogenetic tree builders use a Bayesian approach, as opposed to either UPGMA or NJ. Two of the most popular are Mr. Bayes and BEAST. A comparison of these algorithms would be very interesting, and could be run on sequences from a diverse range of species.
Population Genetics. Many of the datasets above could be used in a population genetics project. You could use deviations from neutrality to detect population size changes or genes under natural selection (let me know if you would like papers related to these topics). You could also try running tree methods on individuals from the same population (i.e. UPGMA) to detect subpopulations and migration events in evolutionary history. Running UPGMA in a sliding window across the genome (to avoid recombination breakpoints) and then aggregating the resulting trees to make evolutionary inferences would make an excellent project (RECOMMENDED).
Hidden Markov Models (HMMs). In class we will study an HMM model for detecting population size changes called PSMC. This is a very elegant method with a software package that is relatively easy to use and often produces very nice results. PSMC can be run on individuals from almost any species with a decent reference genome.
PCA. Running PCA on a dataset (like we will do in Lab 9) is probably not enough for the entire project, but if you would like to incorporate PCA as part of your analysis, you're welcome to do so. It could be an entire project if you work to create a novel dataset and run PCA on that. For example, you could create a multiple sequence alignment (MSA) between humans, Neandertals, and other great apes, use the MSA to create a matrix of SNPs, then run PCA on that. Another option is to post-process the PCA results to try to link them with migrations and splits over time.
Note: I am also open to a more theoretical/mathematical project that does not involve data (population
genetics or HMMs might work well for this). If you have a concrete idea/plan for such a project, let me know.
I would recommend submitting your proposal as soon as possible so you can get feedback and get started. I'll provide feedback on your proposal and create a github repository for your project. The repository should include all the code you write for the project. Only include data if the datasets is very small (i.e. a fraction of the data or test example). The repository should also include your "lab notebook" (see details below) and eventually your slides for the presentation. Summary of what should be on git:
You and your partner should meet with me at least once outside of lab (before or after you submit the proposal, or both). There will also be project check-ins during the last two weeks of lab.
Instead of a formal writeup, you should keep a lab notebook throughout the project (beginning after your git repo is created, but you're welcome to start now if you like). I actually keep an online lab notebook for all my research projects. It helps me remember what I did last and also tracks the time I'm spending on each project. I also use this same document to keep a list of TODOs.
You and your partners should share a lab notebook so you can keep each other updated about your progress. I'm not going to grade this part formally or read every entry - just check that it's there and looks reasonable. It is more for you to keep track of the project.
Here is a recent entry from my own work:
Sara: 03-07-18 (2hrs)