In this lab you will implement your own de Bruijn graph assembler and apply it to a realistic (although synthetic) dataset. The goals of this lab are to develop a deeper understanding of de Bruijn graphs, gain experience going from theory to application, and understand the limitations of our current assembly tools.
Your program should be broken into two files. The first, debruijn.py will contain class definitions for your de Bruijn graph data structure. The second, assembly.py will be your main program that will orchestrate building, traversing, and interpreting the de Bruijn graph.
Find your partner and clone your lab02 git repo as usual. You should see the starter program files, as well as some example datasets. The first step is to construct a de Bruijn graph for a set of reads (in the FASTA file format, same as Lab 1). After creating the de Bruijn graph, we will traverse it to create contigs, which can then be written to a file. Some examples of the program execution are shown below to help you get an idea of what your program should do. They are explained in more detail below.
Here is some output from test1.fasta with k=3:
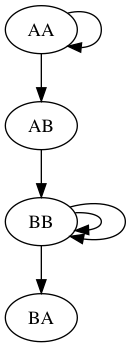
$ python3 assembly.py -r data/test1.fasta -k 3 ---------------------------------------- Welcome to the de Bruijn graph assembler ---------------------------------------- Creating de Bruijn graph with k=3...DONE Display text version of graph (y/n): y Nodes: AA,AB,BB,BA Edges: AA: ['AA', 'AB'] AB: ['BB'] BB: ['BB', 'BB', 'BA'] Write graph visualization to file (y/n): y Enter filename prefix: figs/test1 Determine whether graph is Eulerian (y/n): y Graph Eulerian? True Traverse graph and find contigs (y/n): y Write contigs to file (y/n): y Enter contig filename: results/test1_contigs.fasta Starting path 1/1 from AA: >contig_0 AAABBBBA
In the file figs/test1.png there is a visualization of the graph. It should look like this:
Here is some output from test2.fasta with k=5:
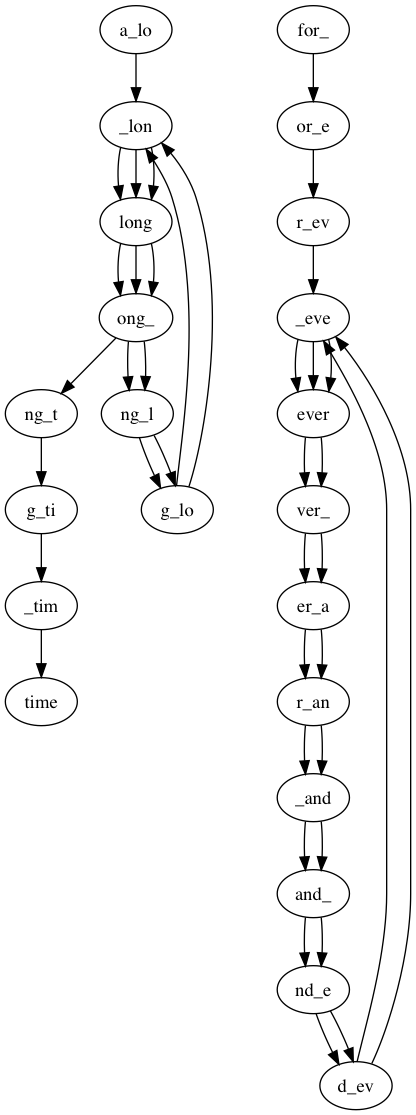
$ python3 assembly.py -r data/test2.fasta -k 5 ---------------------------------------- Welcome to the de Bruijn graph assembler ---------------------------------------- Creating de Bruijn graph with k=5...DONE Display text version of graph (y/n): y Nodes: a_lo,_lon,long,ong_,ng_l,g_lo,ng_t,g_ti,_tim,time,for_,or_e,r_ev,_eve,ever,ver_,er_a,r_an,_and,and_,nd_e,d_ev Edges: a_lo: ['_lon'] _lon: ['long', 'long', 'long'] long: ['ong_', 'ong_', 'ong_'] ong_: ['ng_l', 'ng_l', 'ng_t'] ng_l: ['g_lo', 'g_lo'] g_lo: ['_lon', '_lon'] ng_t: ['g_ti'] g_ti: ['_tim'] _tim: ['time'] for_: ['or_e'] or_e: ['r_ev'] r_ev: ['_eve'] _eve: ['ever', 'ever', 'ever'] ever: ['ver_', 'ver_'] ver_: ['er_a', 'er_a'] er_a: ['r_an', 'r_an'] r_an: ['_and', '_and'] _and: ['and_', 'and_'] and_: ['nd_e', 'nd_e'] nd_e: ['d_ev', 'd_ev'] d_ev: ['_eve', '_eve'] Write graph visualization to file (y/n): y Enter filename prefix: figs/test2 Determine whether graph is Eulerian (y/n): y Graph Eulerian? False Traverse graph and find contigs (y/n): y Write contigs to file (y/n): y Enter contig filename: results/test2_contigs.fasta Starting path 1/2 from a_lo: >contig_0 a_long_long_long_time Starting path 2/2 from for_: >contig_1 for_ever_and_ever_and_ever
In the file figs/test2.png there is a visualization of the graph. It should look like this:
In the file debruijn.py you will define your de Bruijn graph data structure. At minimum, you should have a class for the de Bruijn graph (i.e. DBG) and a Node class for the (k-1)-mer nodes. The methods below provide some guidance, but as long as you're implementing a de Bruijn graph you're welcome to make some reasonable modifications.
Note: you must have some testing in the main function in debruijn.py. I would recommend using test1.fasta for this part. Use if __name__ == "__main__": to prevent this testing from being executed when you import these classes into assembly.py. Make use of assert to ensure you're getting the results you expect in your tests.
First create a Node class (this will be used as part of your DBG class). You are welcome to add more to this class, but at a minimum it should include:
An instance variable for the (k-1)-mer. In essence these nodes are really just wrappers around (k-1)-mers (strings), but with the hash function overridden.
A hash function. We don't want to be comparing (k-1)-mers directly when we're checking whether a new (k-1)-mer is in the graph or not. To make comparisons fast, we will override the hash function. To do this, implement the method __hash__ for this class, which should return the hash of the (k-1)-mer. We'll use the built-in Python hash function: hash(<str>). If you want to assemble the E.coli genome as part of the challenge question, you could think about modifying the hash function to be more tailored to our application.
A __str__ method. For testing purposes, create a string method that returns the (k-1)-mer.
In conjunction with your Node class, create a DBG class with the following functionality. Note that the reads should be read in from a FASTA file. All the reads in the FASTA file will be part of the same de Bruijn graph, which will (hopefully) be connected across reads.
A constructor and the ability to add nodes and edges. There are two main options for this part. Option 1) Have the constructor take in the FASTA filename (i.e. test1.fasta) and read the file in the constructor. Option 2) Have the constructor set up empty node/edge data structures, then read the file in main and call methods for adding nodes/edges as you read the file. Make sure your constructor takes k as a parameter.
Data structures for nodes and edges. This is up to you, but I would recommend having a dictionary of nodes, where the keys are (k-1)-mers (strings) and the values are Node objects (more about this below). For edges, I would recommend having a dictionary where each key is a Node and the value is a list of Nodes it is connected to (outgoing edges).
Method for getting k-mers from a read. You're welcome to make this method private if you like (use __ before the method name).
A __str__ method. At this point, make sure the graph construction is working properly by creating a string method (similar to the program executions shown above). You could also skip to the visualization part (below) before going on to the graph traversals.
A method for determining if the graph is Eulerian. After you have built the de Bruijn graph, the first thing we will check is if it's Eulerian. To do this, we need some way of determining the out-degree and in-degree of each node. We already have the out-degree from the de Bruijn data structure. How can you keep track of the in-degree? However you decide to do this, you should have an Eulerian method that returns True if the graph is Eulerian (at most 2 semi-balanced nodes and all other nodes balanced) and False otherwise. Note that we are not considering different connected components of the graph (although you are welcome to create more nuanced methods that determine if each connected component is Eulerian on its own).
A method for finding semi-balanced nodes. More specifically, create a method that returns a list of nodes with (outdegree - indegree) = 1. These will form our start nodes and we'll start each new path through the graph from one of these nodes.
A method(s) for traversing the graph. Create a method for traversing the graph, forming an Eulerian path (if possible). For the example datasets, the recursive algorithm discussed in class should be fine, but if you're working on the E.coli dataset you will probably want to implement iterative path-finding. For this method we will need a stack to keep track of the path. In Python we can use lists as stacks, using append to add to the stack and pop to remove and return elements from the end. Note that traversing the graph will remove edges, thus destroying the graph. That is okay for this application. Your baseline traversal algorithm should have the start node (one of the semi-balanced nodes you found in the step above) and the path (which starts out empty) as arguments.
A method for finding all the contigs. You should now be able to start at each "start" node, traverse the path, and then find one contiguous sequence (contig). This can be done however you like (the loop over start nodes could be done in the DBG class or in main). Keep in mind the path is returned in reverse order (which is why we need to pop each node off to learn the sequence). In main there will eventually be an option to write the contigs to a FASTA file.
A method for visualizing your graph. For graph visualization, we will use graphviz. To use this library, include:
import graphviz as gvHere is an example of a small directed graph:
g2 = gv.Digraph(format='svg')
g2.node('A')
g2.node('B')
g2.edge('A', 'B')
g2.render('img/g2')
I would recommend using 'png' for the format (you don't have to push the graphs though, so whatever you prefer is fine). The final image renders to img/g2.svg in this example.
More graphviz examples here. For our application, create a directed graph in this style (with (k-1)-mers as the nodes) so the user can visualize the results.
You will define your main program in assembly.py. At a high level, your main should parse the command line arguments and then allow the user to choose the workflow for their DBG assembly. Below are some details about each part.
Parsing Command Line Arguments
For this program we'll start using the optparse library library for parsing command line arguments. Below is some boiler-plate code I use for almost every script I write in Python. It allows you to specify command line arguments with flags so the order of arguments does not matter.
import optparse
import sys
parser = optparse.OptionParser(description='program description')
parser.add_option('-a', '--arg1', type='string', help='helpful message 1')
parser.add_option('-b', '--arg2', type='int', help='helpful message 2')
(opts, args) = parser.parse_args()
# you can include any subset of arguments as mandatory
# (if an argument is not mandatory, it should have a default)
mandatories = ['arg1', 'arg2']
for m in mandatories:
if not opts.__dict__[m]:
print('mandatory option ' + m + ' is missing\n')
parser.print_help()
sys.exit()
# later on in your code...
print(opts.arg1, opts.arg2)For this program you should have at least -r for the FASTA file of reads and -k for the k-mer length. If you include other arguments, make sure to document them. After getting the command line arguments, you can use them with opts.arg1, etc. Make sure to include more helpful names than arg1 and arg2.
Assembly Workflow
After parsing the arguments and building the de Bruijn graph, ask the user several questions about what they would like to do as the graph is analyzed and traversed. For all of these questions, y means yes and anything else means no (so no need to do extensive checking of user input). The reason we don't do all these steps automatically is that for large graphs, it may not be desirable to print or visualize the entire graph. And for small graphs, we may not feel the need to write the contigs to a file explicitly. Note that these steps are only offered once per execution of the program (no need to have a loop unless you want to).
Here are the specific details, but feel to add more functionality or print additional information along the way (in particular, you might want to print out the number of balanced and semi-balanced nodes).
Greet the user.
Create the de Bruijn graph with the given k value and print a message when finished.
Ask the user if they would like to print a text version of the graph. If yes, call the __str__ method for the DBG using print(..).
Ask the user if they would like a visualization written to a file. If yes, prompt them for a filename prefix and then create the visualization (using graphviz).
Ask the user if they would like to determine if the graph is Eulerian. If yes, print out True or False. You can also print out additional information like the number of balanced and semi-balanced nodes.
Ask the user if they would like to traverse the graph and find contigs. If yes, ask them if they would like to write the contigs to a file. If yes, ask them for the filename. Then traverse the graph (starting at each "start" node in turn). If they have requested writing to a file, write the contigs in FASTA format (similar to the example runs above with labels for the name of each contig).
(Optional) Compute and print the N50 of the contigs.
There are 3 test files to practice on: test1.fasta, test2.fasta, and genomeX_reads.fasta. For the third one, the reference (original) genome is provided as well: genomeX_reference.fasta. To see how closely your assembly matches the original, you can use the online tool BLAST and upload the two FASTA files. In the next few weeks we will see how to do this step (called sequence alignment) ourselves.
It is okay if your assembly for the last test case has some issues, the coverage is good but not completely uniform. Experiment with k. In your README, explain which value of k produced the best assembly (there are a few questions to answer about your best assembly). You should also push the contigs for your best k value for this example.
In addition to the requirements listed above, you should ensure your code satisfies these general guidelines:
debruijn.py file.This extension ask you to analyze the runtime of building a de Bruijn graph. For this I would recommend using the E.coli reads and vary the number of reads you consider. Then plot the runtime as a function of the number of reads, in runtime.py. I will show the best runtime visualizations in class. Here is a rough sketch of how to use matplotlib for this purpose. I am happy to help get this working further.
import matplotlib.pyplot as plt
x = range(10)
y = [val**2 for val in x]
plt.plot(x,y,'bo-') # make sure x and y are the same length
plt.title("My Plot")
plt.xlabel("x-axis label")
plt.ylabel("y-axis label")
plt.legend(["line 1"])
plt.show()
I have also provided a set of reads from the E.coli genome from last time: /home/smathieson/public/cs68/ecoli_reads.fasta. The goal is to assemble them into the best possible contigs (the reference is available to you so you can check your solution with BLAST as you go). Any algorithm is fine as long as it is something you come up with (i.e. you cannot just run the reads through Velvet!) Feel free to use literature such as the Velvet paper for inspiration, just not any one else's code (that applies for the rest of the lab as well). If you attempt this part, include your solution in challenge.py, which could also make use of data structures (new or ones you had before) in debruijn.py.
For the challenge, make it clear in your README how I should run your code, which parameters, etc. But don't push your contigs since they will likely be quite large. There will be a prize for the group with the best assembly :)
Make sure to fill out the README file (including some questions about your best choice for k for the third test case). Make sure to also push your best assembly for genomeX. The files you should have at the end are:
README.mddebruijn.pyassembly.pygenomeX_contigs.fastaruntime.py + plots (optional)challenge.py (optional)