CSC 111: Intro to Computer Science through Programming
Lab 10
Due: Friday, Apr. 21 at 11:59pm on Moodle
For this lab, first find your randomly assigned partner. Introduce yourselves - the person with the first name that comes first alphabetically should begin as the "driver", with the other partner as the "navigator". The driver will have the code open, and the navigator will have these instructions open.
At the end of the lab, email the code (finished or not) and transcript to the person who started as the navigator. If you do not finish during lab you have two options:
(1) Arrange to meet before Friday and finish the lab together.
(2) Continue the code separately and denote the part you did on your own with a comment.
Note: it is not an option for one person to complete the code on their own and then send the finished code to their partner to submit. Any code that you submit should be either written by you, or written by you and your partner while you were pair programming. Both partners should submit their code on Moodle.
Classes in Biology
In this lab we will be analyzing DNA information from several different species, contained in the file DNA_data.txt. Note that this is synthetic data, but loosely based on the data in Joe Felsenstein's book: "Inferring Phylogenies". First investigate the file. Each line contains the species name first, followed by their DNA sequence (a very small portion). The end goal of this lab is to find out how many differences there are between each pair of species. This type of computation is very important for creating phylogenetic trees, which model evolution over time. An example is shown below:
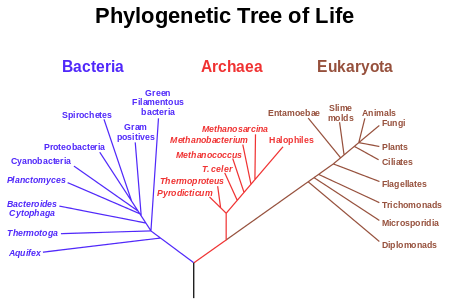
Credit: wikipedia
The steps below will help you develop a program to compute pairwise differences.
- Genome constructor and getters
First create a new class called Genome. This class will represent the information for the genome of a single species. Each instance of this class should know two things: their species name (string) and their DNA sequence (also a string). Based on this information, write the constructor for this class (it will be a lot shorter than our graphics constructors - no need to do anything complicated!) In addition to the constructor, write two getter methods for each instance variable:
- get_name(): which should return the species name
- get_sequence(): which should return the sequence of DNA
- get_name(): which should return the species name
- Read the DNA file
After you have written the constructor, start your main function. The first step in main will be to read the file of DNA information and construct a list of Genomes. This is very similar to Homework 9 with the fish, except that we will need to read a file as well. At the end of this step, you should have a list of Genomes (just one list, not two separate lists of the names and the sequences). Store the length of this list as a variable, and print the variable to make sure it is equal to 8.
Notes: when viewing the file it looks like the name and the sequence are on separate lines, but they are actually on the same line. Make sure to use split(..) to separate the name from the sequence. The loop you use to read the file should be same loop where you call the genome constructor.
After this step you should be able to do the following test:
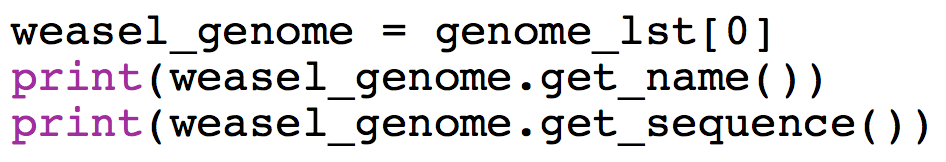
Switch Driver and Navigator here!
- differences(..) method
Next we will write a key method in the Genome class: differences(..). This method will take in one non-self parameter, another instance of the Genome class. The goal of this method is to determine the number of differences between the sequence of the given genome (represented by "self") and the sequence of another genome (often called "other").
This code will be very similar to Lab 3, Part E, except without dealing with missing data. Make sure to return the number of differences at the end of the method. At the end of this step, you should be able to perform the following tests in main, with results 2 and 10:

Optional assert step (skip if you are low on time, we'll talk more about assert later)To provide a check on the input, first check that the sequences are of the same length. One way to do this is to use assert. Assert is a keyword, and the expression after assert should evaluate to a boolean. If the boolean is True, the assertion holds and nothing happens. If the boolean is False, the assertion fails and an error is thrown. This is a useful debugging tool because it checks the input before trying out a more complex computation. Try out the example in the shell below:

Incorporate an assertion into your method to check the sequences are the same length.
- Matrix class
Now we are ready to use this method on all pairs of DNA sequences. We will store the results of these comparisons in a 2D list, or "list of lists", or matrix. We can think about this data structure as an Excel spreadsheet or another type of grid of information. The entry in the ith row and the jth column will be the number of differences between the ith species and the jth species.
In the shell, experiment with the following code, which will add on 4 lists of 4 zeros each using a loop:

Now change one of the entries of the matrix:
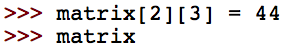
Does the result make sense? To practice classes and encapsulating data, we will make a Matrix class. This class should contain the following methods:
- Constructor: the constructor should take in the number of rows and the number of columns that the matrix should contain. In the constructor, create a new empty matrix of all zeros, called self.data. This is the most important instance variable for the class (I don't think you will need any others, but feel free to use more if it is useful for you).
- A get_entry(i,j) method: This method should return the entry in the ith row and jth column of the matrix.
- A set_entry(i,j,value) method: This method should modify the matrix data in the ith row and jth column to be equal to the parameter value.
- A print() method: For now this method will just print your instance variable, the matrix data. At the end you will modify this method to make the printing more readable.

Switch Driver and Navigator here!
- Constructor: the constructor should take in the number of rows and the number of columns that the matrix should contain. In the constructor, create a new empty matrix of all zeros, called self.data. This is the most important instance variable for the class (I don't think you will need any others, but feel free to use more if it is useful for you).
- Fill the matrix
Now we are ready to fill the matrix with non-zero numbers. In main, use a nested for-loop to iterate twice over the list of genomes. Use the two for-loop indices to obtain a pair of genomes. Then call the differences method, using one genome as the instance and the other as the parameter. After obtaining the number of differences from this method, modify the matrix using the set_entry(...) method to store the result.
When you are done with this step you should see a completed matrix. What are the numbers down the diagonal of this matrix? Does that make sense? Here is what you should get for the first row of this matrix:
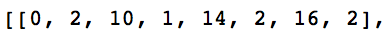
Switch Driver and Navigator here!
- Print the matrix
To make our result a bit more interpretable, we will print the matrix. There are two options for this part:
- Option 1 (recommended): Have your the printing step as a modification to your print() method inside the Matrix class, where you pass in a list of the species names (write a getter to obtain them from the Genome class).
- Option 2: Create a function that will take in two arguments: the list of genomes and the matrix of pairwise differences.
Edit: it is now optional to include the species names in the printing. So you can print only the numerical data (one row at a time, even if it has commas and no tabs).

First print each species name, which will require writing a getter inside your genome class. Between each name, use a tab character (special character "\t"). This will keep all the numbers aligned.
Then for each row, print the species name first, followed by the elements of the matrix, also including tabs between them so everything is aligned.
- Option 1 (recommended): Have your the printing step as a modification to your print() method inside the Matrix class, where you pass in a list of the species names (write a getter to obtain them from the Genome class).
- Analysis
Instead of a transcript, copy over just your formatted matrix into a plain text file (it should preserve the formatting). Then answer the three questions below in this txt file:
- Why are there zeros along the diagonal of the matrix?
- Which two species are the most similar? (note that this is a very small section of the genome, so the number of differences is artificially low)
- Which two species are the most different?
- Why are there zeros along the diagonal of the matrix?
Extensions
- Instead of just looking to determine the min/max number of differences, write separate min/max methods for this type of matrix (make sure to avoid the diagonal when determining the minimum).
- Based on this matrix, what is your guess at the phylogenetic tree relating these species? Try to draw it out. (Note: this is a very hard problem, but we can gain some intuition from the pairwise differences matrix).
Submit
Make sure that both you and your partner have a copy of all the code written during the lab period. Both partners should submit the files:
- lab10.py
- analysis.txt